
Tumor evolution
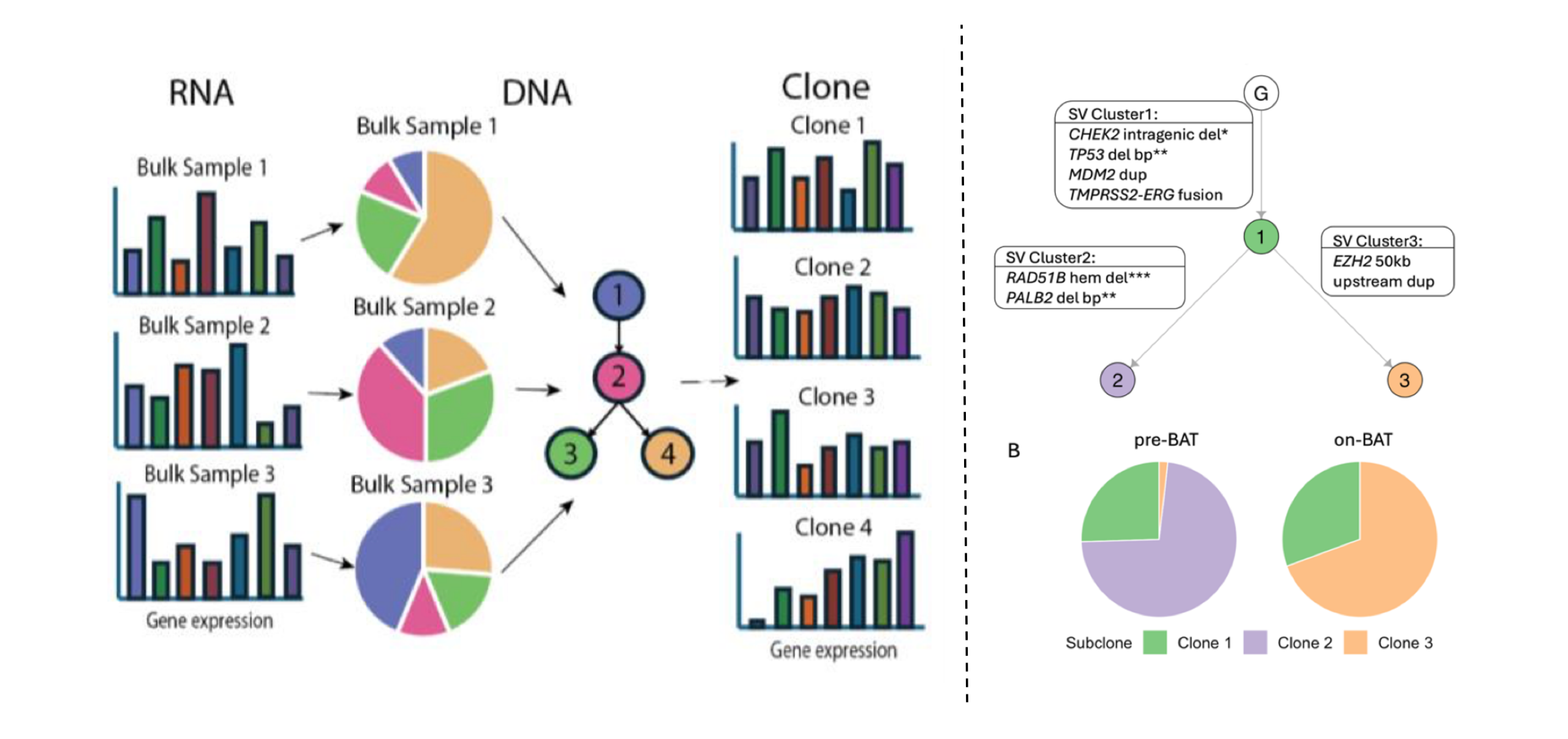 We develop algorithms to reconstruct tumor clonal architectures and evolutionary trees from somatic alterations across single tumors or multiple spatial/longitudinal samples. Our reconstructions model clonal architecture and estimate subclone proportions in each sample, allowing us to track lineages that expand or contract over space and time; the most expanded subclones are expected to have the highest fitness, including treatment-resistant clones that are positively selected under therapy.
We develop algorithms to reconstruct tumor clonal architectures and evolutionary trees from somatic alterations across single tumors or multiple spatial/longitudinal samples. Our reconstructions model clonal architecture and estimate subclone proportions in each sample, allowing us to track lineages that expand or contract over space and time; the most expanded subclones are expected to have the highest fitness, including treatment-resistant clones that are positively selected under therapy.
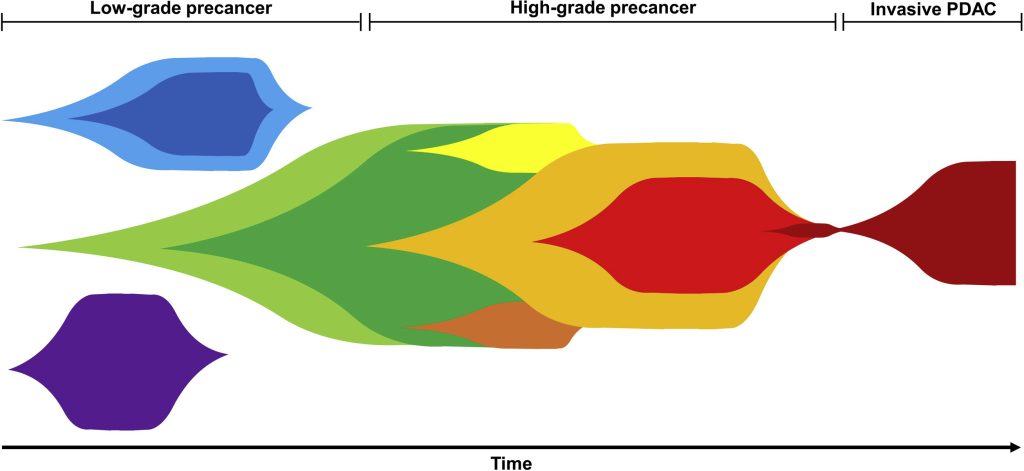
In 2015, we introduced SCHISM (Subclonal Hierarchy Inference of Somatic Mutations), which established a hypothesis-testing framework to determine whether prevalence differences between putative ancestral and descendant clones are statistically significant, and paired this with a genetic algorithm to efficiently explore the space of candidate evolutionary trees. In high-grade serous ovarian cancer (HGSOC), applying this method provided the first genetic evidence supporting a fallopian tube origin for this disease. SCHISM primarily advances tree inference (test + GA search) given known cellular prevalence inputs, whereas PICTograph (2022) advances end-to-end inference by introducing a Bayesian generative clustering model and using sample-presence constraints to sharply prune, and often near-exhaustively score, the plausible tree space (still leveraging SCHISM’s scoring at the end). In pancreatic cancer (PDAC) precursor lesions, PICTograph found evidence of multiclonal initiation by competing driver events, later supported by single-cell DNA profiling. SVCFit (2026) now extends clonal inference to structural variants, enabling SV-informed phylogenies that capture therapy-driven clonal turnover; in metastatic prostate cancer treated with bipolar androgen therapy, SV-defined lineages, modeled by SVCFit, revealed contraction of highly rearranged clones and expansion of resistant subclones associated with specific SV events (e.g., FOXA1 locus remodeling and an EZH2 regulatory-region duplication). PICTographPlus (2026) further integrates DNA phylogenies with bulk RNA-seq to infer clone-resolved expression programs, mapping transcriptional shifts to specific evolutionary branches; in NSCLC and pancreatic cancer, it has linked progression and metastasis to branch-specific programs (e.g., SMAD4-loss versus HR-impairment/EMT trajectories and organ-adapted liver metastatic signatures).
We are now developing methods that incorporate cell–cell associations inferred from single-cell and spatial transcriptomics to connect clonal evolution with tumor–microenvironment interactions and spatially organized cellular programs.